
RESEARCH
CELL CYCLE QUIESCENCE


During development, cells enter and exit quiescence in response to developmental cues or nutrient signaling. Failure of cells to quiesce results in tumorogenesis, whereas premature quiescence establishment can lead to developmental arrest.
Most prior work has focused on the G0 quiescent state that is established after cells exit mitosis. However, certain stem cell populations quiesce at the G2 stage of the cell cycle, right after the completion of DNA replication. But despite its emerging prevalence across a wide evolutionary spectrum, the mechanisms that allow for the establishment of G2 quiescence and how cells exit the G2 quiescence state to resume proliferation remain understudied.
The nascent C. elegans germline presents a unique opportunity to study G2 quiescence in the context of a physiologically regulated tissue. In C. elegans, two germ cell precursors are born during embryogenesis, at about the 100-cell stage, replicate their DNA and then enter a quiescent G2 state for the remainder of embryogenesis. After hatching, these precursors remain quiescent until the larva gains access to food, after which germ cells enter mitosis and divide.
Using a combination of cell biology, proteomics, genetic screens and developmental analyses we are aiming to answer three key questions:
1. What mechanisms control entry into G2 quiescence?
2. How does external signaling regulate exit from G2 quiescence?
3. How do these signals interface with the cell cycle?
MITOSIS AND EMBRYONIC DEVELOPMENT
C. elegans zygote

GFP::tubulin, mCherry::Histone
C. elegans embryo development
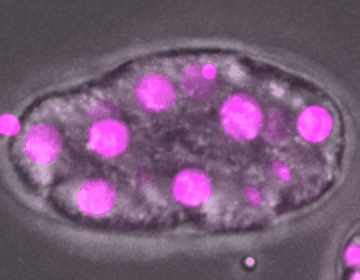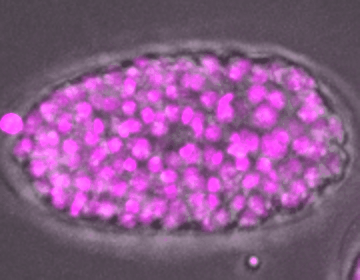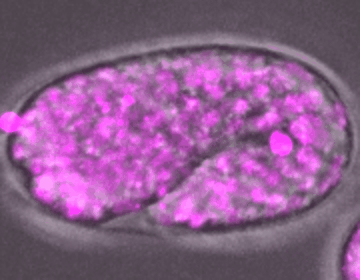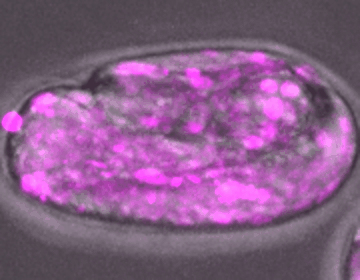
DIC, mCherry::Histone
The development of an embryo depends on the fertilization of an oocyte, which undergoes multiple rounds of mitotic divisions, differentiation, and tissue formation, eventually leading to a fully developed organism. Unraveling the complex signaling networks that coordinate cell division and differentiation during early embryogenesis is crucial to our understanding of early developmental disorders, particularly those of unknown etiology.
Interestingly, the specialization of the cell cycle machinery in embryogenesis is crucial for normal development. The mitotic machinery is essential for events that lead to the specification of cell fate determinants critical for cell differentiation, such as those controlling spindle orientation and asymmetric cell division. Moreover, it plays a crucial role in the expression of cell fate determinants during early embryogenesis by controlling early embryonic translation. The C. elegans embryo, with its fast development time and its conserved molecular pathways, serves as a perfect model to answer these questions.
Using a combination of biochemistry, genetics and live-cell imaging we are working on understanding:
1. What mechanisms ensure rapid mitotic divisions during embryogenesis?
1. How do mitotic control mechanisms integrate with the embryonic development program?
2. What is the importance of mitotic timing control in development?
MITOSIS AND CANCER
Human mitotic cell

Ndc80, ACA
Effect of spindle checkpoint inactivation in human cells

SiR-DNA
Mitosis is an anti-cancer target. Indeed, classical chemotherapy drugs interfere with microtubule dynamics, causing cancer cells to either arrest in mitosis or exit with massive chromosome segregation errors, both of which result in cell death by apoptosis. However, these so-called anti-mitotic drugs indiscriminately target all dividing cells, including stem cells in epithelia and bone marrow. Thus, there is a need to develop novel anti-mitotic cancer therapies with reduced side effects by exploiting cancer-specific mitotic vulnerabilities. Interestingly, cancer cells face several mitotic challenges due to their whole genome duplication and/or pathway missregulation, which often result in chromosomal instability. These challenges present unique opportunities for targeting cancer-specific mitotic mechanisms.
Inspired by recent work translating findings from C. elegans to human cultured cells, we are investigating differences between normal and cancer cells in terms of their mitotic regulation. Our goal is to gain insights into how mitotic pathways differentially control cancer cell division, with the long-term aim of facilitating the development of more specific anti-mitotic drugs with reduced toxicity profiles.
Using a combination of cell biology, proteomics, live imaging microscopy and CRISPR-Cas9 screens, we aim to understand:
1. Are there differences in mitotic timing control between transformed and non-transformed cells?
2. Can we exploit novel mitotic vulnerabilities to selectively target cancer cells?